Sequencing the last 8% of the human genome has taken 20 years and the invention of new techniques for reading long sequences of the genetic code, which consists of the nucleotides C, T, G and A. The entire genome consists of more than 3 billion nucleotides. Credit: Ernesto del Aguila III, NHGRI
DNA, or deoxyribonucleic acid, is a molecule composed of two long strands of nucleotides that coil around each other to form a double helix. It is the hereditary material in humans and almost all other organisms that carries genetic instructions for development, functioning, growth, and reproduction. Nearly every cell in a person’s body has the same DNA. Most DNA is located in the cell nucleus (where it is called nuclear DNA), but a small amount of DNA can also be found in the mitochondria (where it is called mitochondrial DNA or mtDNA).
” data-gt-translate-attributes=”["attribute":"data-cmtooltip", "format":"html"]”>DNA sequences around centromere show history of human genetic variation.
When scientists announced the complete sequence of the human genome in 2003, they were fudging a bit.
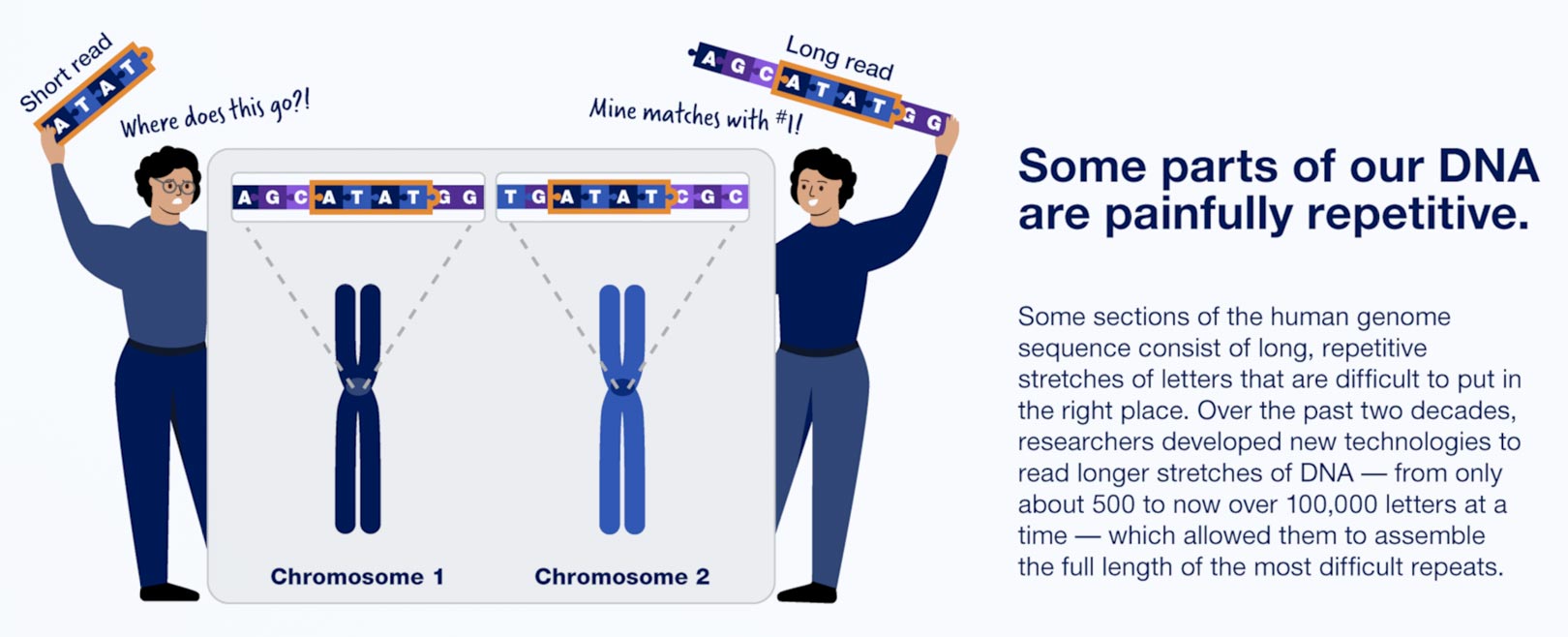
In fact, nearly 20 years later, about 8% of the genome had never been fully sequenced, largely because it consists of highly repetitive chunks of DNA that are hard to align with the rest.
But a three-year-old consortium has finally filled in that remaining DNA, providing the first complete, gapless genome sequence for scientists and physicians to refer to.
The newly completed genome, dubbed T2T-CHM13, represents a major upgrade from the current reference genome, called GRCh38, which is used by doctors when searching for mutations linked to disease, as well as by scientists looking at the evolution of human genetic variation.
Among other things, the new DNA sequences reveal never-before-seen detail about the region around the centromere, which is where chromosomes are grabbed and pulled apart when cells divide, ensuring that each “daughter” cell inherits the correct number of chromosomes. Variability within this region may also provide new evidence of how our human ancestors evolved in Africa.
“Uncovering the complete sequence of these formerly missing regions of the genome told us so much about how they’re organized, which was totally unknown for many chromosomes,” said Nicolas Altemose, a postdoctoral fellow at the <span class="glossaryLink" aria-describedby="tt" data-cmtooltip="
University of California, Berkeley
Located in Berkeley, California and founded in 1868, University of California, Berkeley is a public research university that also goes by UC Berkeley, Berkeley, California, or Cal. It maintains close relationships with three DOE National Laboratories: Lawrence Berkeley National Laboratory, Los Alamos National Laboratory, and Lawrence Livermore National Laboratory.
” data-gt-translate-attributes=”["attribute":"data-cmtooltip", "format":"html"]”>University of California, Berkeley, and a co-author of four new papers about the completed genome. “Before, we just had the blurriest picture of what was there, and now it’s crystal clear down to single base pair resolution.”
Altemose is first author of one paper that describes the base pair sequences around the centromere. A paper explaining how the sequencing was done will appear in the April 1 print edition of the journal Science, while Altemose’s centromere paper and four others describing what the new sequences tell us are summarized in the journal with the full papers posted online. Four companion papers, including one for which Altemose is co-first author, also will appear online April 1 in the journal Nature Methods.
The sequencing and analysis were performed by a team of more than 100 people, the so-called Telemere-to-Telomere Consortium, or T2T, named for the telomeres that cap the ends of all chromosomes. The consortium’s gapless version of all 22 autosomes and the X sex chromosome is composed of 3.055 billion base pairs, the units from which chromosomes and our genes are built, and 19,969 protein-coding genes. Of the protein-coding genes, the T2T team found about 2,000 new ones, most of them disabled, but 115 of which may still be expressed. They also found about 2 million additional variants in the human genome, 622 of which occur in medically relevant genes.
“In the future, when someone has their genome sequenced, we will be able to identify all of the variants in their DNA and use that information to better guide their health care,” said Adam Phillippy, one of the leaders of T2T and a senior investigator at the National Human Genome Research Institute (NHGRI) of the National Institutes of Health. “Truly finishing the human genome sequence was like putting on a new pair of glasses. Now that we can clearly see everything, we are one step closer to understanding what it all means.”
The evolving centromere
The new DNA sequences in and around the centromere total about 6.2% of the entire genome, or nearly 190 million base pairs, or nucleotides. Of the remaining newly added sequences, most are found around the telomeres at the end of each chromosome and in the regions surrounding ribosomal genes. The entire genome is made of just four types of nucleotides, which, in groups of three, code for the <span class="glossaryLink" aria-describedby="tt" data-cmtooltip="
amino acids
<div class="cell text-container large-6 small-order-0 large-order-1">
<div class="text-wrapper"><br />Amino acids are a set of organic compounds used to build proteins. There are about 500 naturally occurring known amino acids, though only 20 appear in the genetic code. Proteins consist of one or more chains of amino acids called polypeptides. The sequence of the amino acid chain causes the polypeptide to fold into a shape that is biologically active. The amino acid sequences of proteins are encoded in the genes. Nine proteinogenic amino acids are called "essential" for humans because they cannot be produced from other compounds by the human body and so must be taken in as food.<br /></div>
</div>
” data-gt-translate-attributes=”["attribute":"data-cmtooltip", "format":"html"]”>amino acids used to build proteins. Altemose’s main research involves finding and exploring areas of the chromosomes where proteins interact with DNA.
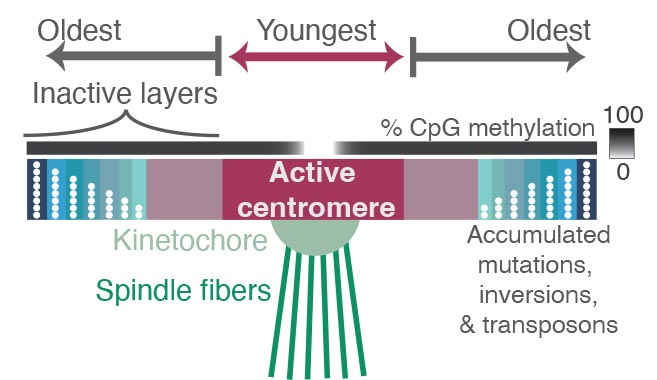
The spindles (green) that pull chromosomes apart during cell division are attached to a protein complex called the kinetochore, which latches onto the chromosome at a place called the centromere — a region containing highly repetitive DNA sequences. Comparing the sequences of these repeats revealed where mutations have accumulated over millions of years, reflecting the relative age of each repeat. Repeats in the active centromere tend to be the youngest and most recently duplicated sequences in the region, and they have strikingly low DNA methylation. Surrounding the active centromere on both sides are older repeats, probably the relics of former centromeres, with the oldest ones farthest from the active centromere. The researchers hope that new experimental methods will help reveal why centromeres evolve from the middle, as well as why this pattern is so closely associated with binding by the kinetochore and with low DNA methylation. Credit: Nicolas Altemose, UC Berkeley
“Without proteins, DNA is nothing,” said Altemose, who earned a Ph.D. in bioengineering jointly from UC Berkeley and UC San Francisco in 2021 after having received a D.Phil. in statistics from Oxford University. “DNA is a set of instructions with no one to read it if it doesn’t have proteins around to organize it, regulate it, repair it when it’s damaged and replicate it. Protein-DNA interactions are really where all the action is happening for genome regulation, and being able to map where certain proteins bind to the genome is really important for understanding their function.”
After the T2T consortium sequenced the missing DNA, Altemose and his team used new techniques to find the place within the centromere where a big protein complex called the kinetochore solidly grips the chromosome so that other machines inside the nucleus can pull chromosome pairs apart.
“When this goes wrong, you end up with missegregated chromosomes, and that leads to all kinds of problems,” he said. “If that happens in meiosis, that means you can have chromosomal anomalies leading to spontaneous miscarriage or congenital diseases. If it happens in somatic cells, you can end up with cancer — basically, cells that have massive misregulation.”
What they found in and around the centromeres were layers of new sequences overlaying layers of older sequences, as if through evolution new centromere regions have been laid down repeatedly to bind to the kinetochore. The older regions are characterized by more random mutations and deletions, indicating they’re no longer used by the cell. The newer sequences where the kinetochore binds are much less variable, and also less methylated. The addition of a methyl group is an epigenetic tag that tends to silence genes.
All of the layers in and around the centromere are composed of repetitive lengths of DNA, based on a unit about 171 base pairs long, which is roughly the length of DNA that wraps around a group of proteins to form a nucleosome, keeping the DNA packaged and compact. These 171 base pair units form even larger repeat structures that are duplicated many times in tandem, building up a large region of repetitive sequences around the centromere.
The T2T team focused on only one human genome, obtained from a non-cancerous tumor called a hydatidiform mole, which is essentially a human embryo that rejected the maternal DNA and duplicated its paternal DNA instead. Such embryos die and transform into tumors. But the fact that this mole had two identical copies of the paternal DNA — both with the father’s X chromosome, instead of different DNA from both mother and father — made it easier to sequence.
The researchers also released this week the complete sequence of a Y chromosome from a different source, which took nearly as long to assemble as the rest of the genome combined, Altemose said. The analysis of this new Y chromosome sequence will appear in a future publication.
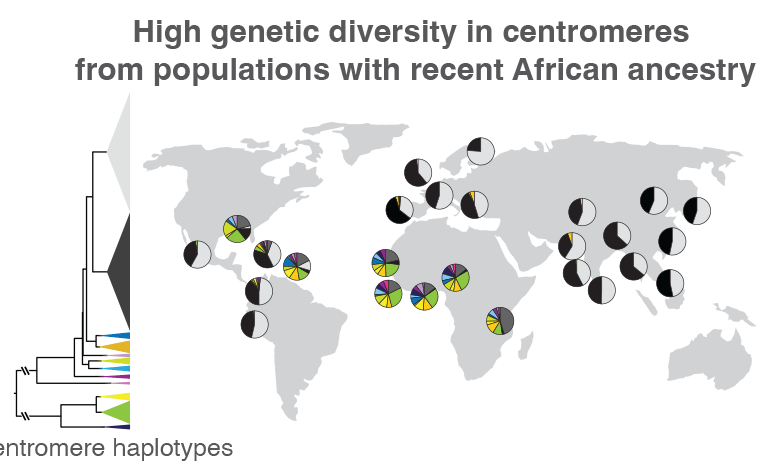
When the researchers compared centromeric regions of 1,600 people from around the world, they found that those without recent African ancestry mostly had two types of sequence variations. The proportions of these two variations are represented by the black and light gray wedges within the circles, which are placed on the map near the location where each group of individuals was sampled. Those from Africa or other areas with a large proportion of people with recent African ancestry, like the Caribbean, had much more centromeric sequence variation, represented by the multi-colored wedges. Such variations could help track how centromeric regions evolve, as well as how these genetic variants are related to health and disease. Credit: Nicolas Altemose, UC Berkeley
Altemose and his team, which included UC Berkeley project scientist Sasha Langley, also used the new reference genome as a scaffold to compare the centromeric DNA of 1,600 individuals from around the world, revealing major differences in both the sequence and copy number of repetitive DNA around the centromere. Previous studies have shown that when groups of ancient humans migrated out of Africa to the rest of the world, they took only a small sample of genetic variants with them. Altemose and his team confirmed that this pattern extends into centromeres.
“What we found is that in individuals with recent ancestry outside the African continent, their centromeres, at least on chromosome X, tend to fall into two big clusters, while most of the interesting variation is in individuals who have recent African ancestry,” Altemose said. “This isn’t entirely a surprise, given what we know about the rest of the genome. But what it suggests is that if we want to look at the interesting variation in these centromeric regions, we really need to have a focused effort to sequence more African genomes and do complete telomere-to-telomere sequence assembly.”
DNA sequences around the centromere could also be used to trace human lineages back to our common ape ancestors, he noted.
“As you move away from the site of the active centromere, you get more and more degraded sequence, to the point where if you go out to the furthest shores of this sea of repetitive sequences, you start to see the ancient centromere that, perhaps, our distant primate ancestors used to bind to the kinetochore,” Altemose said. “It’s almost like layers of fossils.”
Long-read sequencing a game changer
The T2T’s success is due to improved techniques for sequencing long stretches of DNA at once, which helps when determining the order of highly repetitive stretches of DNA. Among these are PacBio’s HiFi sequencing, which can read lengths of more than 20,000 base pairs with high <span class="glossaryLink" aria-describedby="tt" data-cmtooltip="
accuracy
How close the measured value conforms to the correct value.
” data-gt-translate-attributes=”["attribute":"data-cmtooltip", "format":"html"]”>accuracy. Technology developed by Oxford Nanopore Technologies Ltd., on the other hand, can read up to several million base pairs in sequence, though with less fidelity. For comparison, so-called next-generation sequencing by Illumina Inc. is limited to hundreds of base pairs.
One reason it took 20 years to complete the human genome sequence: much of our DNA is extremely repetitive. Credit: Infographic courtesy of NHGRI, NIH
“These new long-read DNA sequencing technologies are just incredible; they’re such game changers, not only for this repetitive DNA world, but because they allow you to sequence single long molecules of DNA,” Altemose said. “You can begin to ask questions at a level of resolution that just wasn’t possible before, not even with short-read sequencing methods.”
Altemose plans to explore the centromeric regions further, using an improved technique he and colleagues at Stanford developed to pinpoint the sites on the chromosome that are bound by proteins, similar to how the kinetochore binds to the centromere. This technique, too, uses long-read sequencing technology. He and his group described the technique, called Directed Methylation with Long-read sequencing (DiMeLo-seq), in a paper that appeared this week in the journal Nature Methods.
Meanwhile, the T2T consortium is partnering with the Human PanGenome Reference Consortium to work toward a reference genome that represents all of humanity.
“Instead of just having one reference from one human individual or one hydatidiform mole, which isn’t even a real human individual, we should have a reference that represents everybody,” Altemose said. “There are various ideas about how to accomplish that. But what we need first is a grasp of what that variation looks like, and we need lots of high-quality individual genome sequences to accomplish that.”
His work on the centromeric regions, which he called “a passion project,” was funded by postdoctoral fellowships. The leaders of the T2T project were Karen Miga of UC Santa Cruz, Evan Eichler of the <span class="glossaryLink" aria-describedby="tt" data-cmtooltip="
University of Washington
Founded in 1861, the University of Washington (UW, simply Washington, or informally U-Dub) is a public research university in Seattle, Washington, with additional campuses in Tacoma and Bothell. Classified as an R1 Doctoral Research University classification under the Carnegie Classification of Institutions of Higher Education, UW is a member of the Association of American Universities.
” data-gt-translate-attributes=”["attribute":"data-cmtooltip", "format":"html"]”>University of Washington, and Adam Phillippy of NHGRI, which provided much of the funding. Other UC Berkeley co-authors of the centromere paper are Aaron Streets, assistant professor of bioengineering; Abby Dernburg and Gary Karpen, professors of molecular and cell biology; project scientist Sasha Langley; and former postdoctoral fellow Gina Caldas.
Reference: “Complete genomic and epigenetic maps of human centromeres” by Nicolas Altemose, Glennis A. Logsdon, Andrey V. Bzikadze, Pragya Sidhwani, Sasha A. Langley, Gina V. Caldas, Savannah J. Hoyt, Lev Uralsky, Fedor D. Ryabov, Colin J. Shew, Michael E. G. Sauria, Matthew Borchers, Ariel Gershman, Alla Mikheenko, Valery A. Shepelev, Tatiana Dvorkina, Olga Kunyavskaya, Mitchell R. Vollger, Arang Rhie, Ann M. McCartney, Mobin Asri, Ryan Lorig-Roach, Kishwar Shafin, Julian K. Lucas, Sergey Aganezov, Daniel Olson, Leonardo Gomes de Lima, Tamara Potapova, Gabrielle A. Hartley, Marina Haukness, Peter Kerpedjiev, Fedor Gusev, Kristof Tigyi, Shelise Brooks, Alice Young, Sergey Nurk, Sergey Koren, Sofie R. Salama, Benedict Paten, Evgeny I. Rogaev, Aaron Streets, Gary H. Karpen, Abby F. Dernburg, Beth A. Sullivan, Aaron F. Straight, Travis J. Wheeler, Jennifer L. Gerton, Evan E. Eichler, Adam M. Phillippy, Winston Timp, Megan Y. Dennis, Rachel J. O’Neill, Justin M. Zook, Michael C. Schatz, Pavel A. Pevzner, Mark Diekhans, Charles H. Langley, Ivan A. Alexandrov and Karen H. Miga, 1 April 2022, Science. DOI: 10.1126/science.abl4178
More than 40 trillion gallons of rain drenched the Southeast United States in the last week from Hurricane Helene and a run-of-the-mill rainstorm that sloshed in ahead of it — an unheard of amount of water that has stunned experts.
That’s enough to fill the Dallas Cowboys’ stadium 51,000 times, or Lake Tahoe just once. If it was concentrated just on the state of North Carolina that much water would be 3.5 feet deep (more than 1 meter). It’s enough to fill more than 60 million Olympic-size swimming pools.
“That’s an astronomical amount of precipitation,” said Ed Clark, head of the National Oceanic and Atmospheric Administration’s National Water Center in Tuscaloosa, Alabama. “I have not seen something in my 25 years of working at the weather service that is this geographically large of an extent and the sheer volume of water that fell from the sky.”
The flood damage from the rain is apocalyptic, meteorologists said. More than 100 people are dead, according to officials.
Private meteorologist Ryan Maue, a former NOAA chief scientist, calculated the amount of rain, using precipitation measurements made in 2.5-mile-by-2.5 mile grids as measured by satellites and ground observations. He came up with 40 trillion gallons through Sunday for the eastern United States, with 20 trillion gallons of that hitting just Georgia, Tennessee, the Carolinas and Florida from Hurricane Helene.
Clark did the calculations independently and said the 40 trillion gallon figure (151 trillion liters) is about right and, if anything, conservative. Maue said maybe 1 to 2 trillion more gallons of rain had fallen, much if it in Virginia, since his calculations.
Clark, who spends much of his work on issues of shrinking western water supplies, said to put the amount of rain in perspective, it’s more than twice the combined amount of water stored by two key Colorado River basin reservoirs: Lake Powell and Lake Mead.
Several meteorologists said this was a combination of two, maybe three storm systems. Before Helene struck, rain had fallen heavily for days because a low pressure system had “cut off” from the jet stream — which moves weather systems along west to east — and stalled over the Southeast. That funneled plenty of warm water from the Gulf of Mexico. And a storm that fell just short of named status parked along North Carolina’s Atlantic coast, dumping as much as 20 inches of rain, said North Carolina state climatologist Kathie Dello.
Then add Helene, one of the largest storms in the last couple decades and one that held plenty of rain because it was young and moved fast before it hit the Appalachians, said University of Albany hurricane expert Kristen Corbosiero.
“It was not just a perfect storm, but it was a combination of multiple storms that that led to the enormous amount of rain,” Maue said. “That collected at high elevation, we’re talking 3,000 to 6000 feet. And when you drop trillions of gallons on a mountain, that has to go down.”
The fact that these storms hit the mountains made everything worse, and not just because of runoff. The interaction between the mountains and the storm systems wrings more moisture out of the air, Clark, Maue and Corbosiero said.
North Carolina weather officials said their top measurement total was 31.33 inches in the tiny town of Busick. Mount Mitchell also got more than 2 feet of rainfall.
Before 2017’s Hurricane Harvey, “I said to our colleagues, you know, I never thought in my career that we would measure rainfall in feet,” Clark said. “And after Harvey, Florence, the more isolated events in eastern Kentucky, portions of South Dakota. We’re seeing events year in and year out where we are measuring rainfall in feet.”
Storms are getting wetter as the climate change s, said Corbosiero and Dello. A basic law of physics says the air holds nearly 4% more moisture for every degree Fahrenheit warmer (7% for every degree Celsius) and the world has warmed more than 2 degrees (1.2 degrees Celsius) since pre-industrial times.
Corbosiero said meteorologists are vigorously debating how much of Helene is due to worsening climate change and how much is random.
For Dello, the “fingerprints of climate change” were clear.
“We’ve seen tropical storm impacts in western North Carolina. But these storms are wetter and these storms are warmer. And there would have been a time when a tropical storm would have been heading toward North Carolina and would have caused some rain and some damage, but not apocalyptic destruction. ”
Associated Press climate and environmental coverage receives support from several private foundations. See more about AP’s climate initiative here. The AP is solely responsible for all content.
It’s a dinosaur that roamed Alberta’s badlands more than 70 million years ago, sporting a big, bumpy, bony head the size of a baby elephant.
On Wednesday, paleontologists near Grande Prairie pulled its 272-kilogram skull from the ground.
They call it “Big Sam.”
The adult Pachyrhinosaurus is the second plant-eating dinosaur to be unearthed from a dense bonebed belonging to a herd that died together on the edge of a valley that now sits 450 kilometres northwest of Edmonton.
It didn’t die alone.
“We have hundreds of juvenile bones in the bonebed, so we know that there are many babies and some adults among all of the big adults,” Emily Bamforth, a paleontologist with the nearby Philip J. Currie Dinosaur Museum, said in an interview on the way to the dig site.
She described the horned Pachyrhinosaurus as “the smaller, older cousin of the triceratops.”
“This species of dinosaur is endemic to the Grand Prairie area, so it’s found here and nowhere else in the world. They are … kind of about the size of an Indian elephant and a rhino,” she added.
The head alone, she said, is about the size of a baby elephant.
The discovery was a long time coming.
The bonebed was first discovered by a high school teacher out for a walk about 50 years ago. It took the teacher a decade to get anyone from southern Alberta to come to take a look.
“At the time, sort of in the ’70s and ’80s, paleontology in northern Alberta was virtually unknown,” said Bamforth.
When paleontogists eventually got to the site, Bamforth said, they learned “it’s actually one of the densest dinosaur bonebeds in North America.”
“It contains about 100 to 300 bones per square metre,” she said.
Paleontologists have been at the site sporadically ever since, combing through bones belonging to turtles, dinosaurs and lizards. Sixteen years ago, they discovered a large skull of an approximately 30-year-old Pachyrhinosaurus, which is now at the museum.
About a year ago, they found the second adult: Big Sam.
Bamforth said both dinosaurs are believed to have been the elders in the herd.
“Their distinguishing feature is that, instead of having a horn on their nose like a triceratops, they had this big, bony bump called a boss. And they have big, bony bumps over their eyes as well,” she said.
“It makes them look a little strange. It’s the one dinosaur that if you find it, it’s the only possible thing it can be.”
The genders of the two adults are unknown.
Bamforth said the extraction was difficult because Big Sam was intertwined in a cluster of about 300 other bones.
The skull was found upside down, “as if the animal was lying on its back,” but was well preserved, she said.
She said the excavation process involved putting plaster on the skull and wooden planks around if for stability. From there, it was lifted out — very carefully — with a crane, and was to be shipped on a trolley to the museum for study.
“I have extracted skulls in the past. This is probably the biggest one I’ve ever done though,” said Bamforth.
“It’s pretty exciting.”
This report by The Canadian Press was first published Sept. 25, 2024.
TEL AVIV, Israel (AP) — A rare Bronze-Era jar accidentally smashed by a 4-year-old visiting a museum was back on display Wednesday after restoration experts were able to carefully piece the artifact back together.
Last month, a family from northern Israel was visiting the museum when their youngest son tipped over the jar, which smashed into pieces.
Alex Geller, the boy’s father, said his son — the youngest of three — is exceptionally curious, and that the moment he heard the crash, “please let that not be my child” was the first thought that raced through his head.
The jar has been on display at the Hecht Museum in Haifa for 35 years. It was one of the only containers of its size and from that period still complete when it was discovered.
The Bronze Age jar is one of many artifacts exhibited out in the open, part of the Hecht Museum’s vision of letting visitors explore history without glass barriers, said Inbal Rivlin, the director of the museum, which is associated with Haifa University in northern Israel.
It was likely used to hold wine or oil, and dates back to between 2200 and 1500 B.C.
Rivlin and the museum decided to turn the moment, which captured international attention, into a teaching moment, inviting the Geller family back for a special visit and hands-on activity to illustrate the restoration process.
Rivlin added that the incident provided a welcome distraction from the ongoing war in Gaza. “Well, he’s just a kid. So I think that somehow it touches the heart of the people in Israel and around the world,“ said Rivlin.
Roee Shafir, a restoration expert at the museum, said the repairs would be fairly simple, as the pieces were from a single, complete jar. Archaeologists often face the more daunting task of sifting through piles of shards from multiple objects and trying to piece them together.
Experts used 3D technology, hi-resolution videos, and special glue to painstakingly reconstruct the large jar.
Less than two weeks after it broke, the jar went back on display at the museum. The gluing process left small hairline cracks, and a few pieces are missing, but the jar’s impressive size remains.
The only noticeable difference in the exhibit was a new sign reading “please don’t touch.”