Sample collection
For reagent testing, we collected ten unmodified faunal remains, similar in and size and shape to material used in artefact production, from the late Middle Palaeolithic and early Upper Palaeolithic deposits of the French sites Quinçay (seven) and Les Cottés (three) (Extended Data Table 1 and Extended Data Fig. 2). The estimated ages of these specimens range from 55 to 35 kyr21,37,38. Non-destructive DNA extraction was then applied to 15 osseous specimens excavated at Quinçay Cave, all from layers attributed to the Châtelperronian technocomplex and probably dating to 45–35 ka21,37, to three tooth pendants excavated in the Initial Upper Palaeolithic (45–43 ka)39 layers in the niche 1 area of Bacho Kiro Cave40,41, as well as on one tooth pendant excavated in 2019 in layer 11 (39–24 ka), square E-3, of the south chamber of Denisova Cave (Extended Data Table 1 and Extended Data Fig. 3). Samples from Bacho Kiro and Denisova Cave were excavated and handled using sterile gloves and additional precautions were taken to minimize the introduction of modern DNA contamination. More information about the samples and the archaeological context in which they were recovered is provided in Supplementary Information 1.
Testing reagents for non-destructive DNA extraction using 3D surface texture measurements
We evaluated four reagents for their potential use in non-destructive DNA extraction by applying each to two Pleistocene samples comparable with the ones usually transformed into bone or tooth tools and ornaments, preferably choosing one bone and one tooth fragment for each reagent (Extended Data Table 1). As none of the objects was perfectly clean and sediment microparticles may be released upon exposure to liquids, we also performed treatments with water to obtain baseline measurements of changes in the microtopography that are independent of the chemical compositions of the reagents. Reagents and incubation conditions were as follows:
- (1)Following a protocol42 further detailing the non-destructive DNA extraction method for museum specimens by Rohland et al. (2004)12, samples were completely submerged in 10–15 ml guanidinium thiocyanate buffer (5 M guanidinium isothiocyanate, 50 mM Tris-HCl, pH 8.0, 25 mM NaCl, 1.3% Triton X-100, 20 mM EDTA, pH 8.0, and 50 mM DTT) and incubated for 5 days in the dark.
- (2)Incubation in EDTA, supplemented with a detergent (0.45 M EDTA, pH 8.0, and 0.05% Tween-20), was performed for 15 min at room temperature. EDTA is widely used in bone extraction protocols as a decalcifying agent to dissolve hydroxyapatite, the main mineral component of bones14,15,16. It is therefore expected to cause severe alterations to the sample material and was included to demonstrate the effect of such alterations on the quantitative 3D-surface texture analysis (3DST) measurements.
- (3)Two samples were submerged for 15 min in 0.5% sodium hypochlorite (bleach) solution, a reagent previously used for decontamination of ancient skeletal remains11,17. Bleach treatment destroys surface-bound DNA and is not suitable for DNA extraction. However, it may be used in later implementations of the method to remove contaminant DNA before non-destructive DNA extraction.
- (4)Temperature-controlled release of DNA was performed following a modified version of the method by Essel et al. (2021)18 by submerging the samples in sodium phosphate buffer (0.5 M sodium phosphate, pH 7.0, and 0.1% Tween-20). Serial incubations were performed at 21 (room temperature), 37, 60 and 90 °C, each with incubation times of 30 min for a total of three incubations per temperature.
All treatments were performed in 50-ml Falcon tubes without agitation to avoid any mechanical damage to the sample. Reagent volumes were chosen individually for each sample (ranging from 5 ml to 17.5 ml) to ensure complete submergence. Incubations above room temperature were performed in a Heating-ThermoMixer MHR 11 (Hettich Benelux) equipped with inserts for 50-ml Falcon tubes. For the 90 °C incubation in phosphate buffer, the temperature of the device was set to 99 °C to ensure that 90 °C was reached inside the tube by the end of the incubation time. After the treatments, all samples were placed in fresh 50-ml Falcon tubes and incubated in water for 1 h at room temperature to remove residual reagents. Samples were dried at room temperature for 5 days before they were returned to their storage containers.
Changes in the microtopography of the bone or tooth objects were tracked using quantitative 3DST19 before and after extraction following established protocols19,43. Meshed axiomatic 3D models were generated and the following ISO 25178 parameters were used for statistical testing (paired Student’s t-test, before and after the treatment, α ≤ 0.01): mean roughness (Sa), void volume (Vvv), peak curvature (Spc) and peak density (Spd). Further details regarding the 3DST measurements are provided in Supplementary Information 2.
In addition, we explored the compatibility of phosphate-based non-destructive DNA extraction with subsequent 14C dating (Supplementary Information 3).
Non-destructive DNA isolation from artefacts
As sodium phosphate buffer did not cause substantial alterations of ancient bones and teeth in 3DST measurements, we modified a previously described method for temperature-controlled gradual DNA release from bone and tooth powder using this reagent18 for non-destructive DNA extraction from complete bones and teeth. Stepwise extraction of DNA makes it possible to closely monitor the release of different DNA components during the extraction process (endogenous DNA, environmental DNA from the surrounding sediment, ancient human DNA and present-day contamination), potentially allowing inferences to be drawn about whether these components originate from traces of sediment that may still be adherent to the object, from its surface or its interior. We then applied this protocol to a total of 15 specimens from Quinçay, Bacho Kiro Cave and Denisova Cave. Temperature-controlled non-destructive DNA extraction was performed in an ancient DNA clean-room at the Max Planck Institute for Evolutionary Anthropology in Leipzig, Germany, according to the four steps below (see Fig. 1a for a schematic overview):
(1) Removal of sediment
This step was only performed for the freshly excavated material. First, clumps of sediment attached to the specimen were carefully removed by hand using a flexible disposable plastic microspatula. The specimen was then put into a 50-ml Falcon tube, rinsed by pouring between 20 ml and 50 ml of water into the tube, and transferred to a fresh Falcon tube. This procedure was repeated two to three times until no more sediment was released into the water. The tubes with water containing the sediment that had been washed off were then centrifuged at 16,400g for 5 min to pellet the sediment, and the clear supernatants were transferred to fresh tubes. All three types of material collected in this procedure (the manually removed sediment, the sediment pellet collected by centrifugation and the clear water) were subsequently subjected to DNA purification (see below).
(2) Temperature-controlled DNA release using sodium phosphate buffer
Each cleaned specimen was put into a 50-ml Falcon tube to which sodium phosphate buffer (0.5 M sodium phosphate, pH 7.0, and 0.1% Tween-20) was added until the specimen was completely submerged in the reagent (between 5 ml and 50 ml). After 30 min of incubation at room temperature (without agitation), the buffer was transferred to a fresh 50-ml Falcon tube. This step was repeated twice at room temperature (21 °C, for a total of three incubations) and then three times each at 37 °C, 60 °C and 90 °C (see above for details on the device and temperature settings used). A final incubation in water was performed at room temperature to remove residual reagent and the specimens were dried at room temperature for 5 days.
(3) DNA concentration
To facilitate subsequent DNA purification, half of the volumes of the water and phosphate buffer DNA fractions generated in steps (1) and (2) were reduced to between 50 µl and 75 µl by concentrating the DNA using Amicon Ultra-4 Centrifugal Filter Units with Ultracel-3 membranes (Millipore). For this, up to 4 ml or 15 ml of the respective sample was added to a filter unit, which was spun for 90 min at 4,000g in a centrifuge with an active cooling unit set to 21 °C. In cases in which the sample volume exceeded 4 ml or 15 ml, the flow-through was discarded and the filter unit was reloaded with remaining sample. Finally, a buffer exchange was performed by adding 4 ml or 15 ml TE buffer (10 mM Tris-HCl, pH 8.0, and 1 mM EDTA) to the concentrated sample on top of the filter unit and spinning for 30 min at 4,000g. The supernatant was filled up to 300 µl with TE buffer (10 mM Tris-HCl, 1 mM EDTA, pH 8.0) and then transferred into a fresh 1.5-ml Eppendorf low-bind tube and stored at −20 °C until further processing.
(4) DNA purification
DNA was isolated from the concentrated DNA fractions prepared in step (3) using a column-based method for silica-based ancient DNA extraction detailed elsewhere44. For this, 300 µl concentrated DNA was used as input for DNA purification using binding buffer ‘D’ and the purified DNA was recovered in 50 µl elution buffer. DNA extraction from the sediment samples removed from the artefacts manually or through water washes (sediment pellets) was performed using the same method but different input volumes. In brief, lysates were prepared by transferring up to 130 mg sediment to a 2-ml low-bind Eppendorf tube, adding up to 2 ml lysis buffer (0.45 M EDTA, pH 8.0, 0.05% Tween-20 and 0.25 mg ml−1 proteinase K) (1 ml for samples of less than 100 mg or 0.5 ml for samples of less than 25 mg) and incubating overnight at 37 °C under rotation. DNA purification was performed using 500 µl or 1,000 µl lysate.
Negative controls containing sodium phosphate buffer or lysis buffer without sample material were carried alongside the samples through all steps of subsequent sample preparation and sequencing. For a subset of samples, DNA was extracted from only the first phosphate buffer fraction produced at each of the four incubation temperatures. Supplementary Data 1 provides an overview of the DNA extracts generated in this study.
Library preparation
Of the extract, 10 µl was then converted into single-stranded DNA libraries using the automated protocol described in Gansauge et al. (2020)23. The number of unique library molecules obtained and the efficiency of library preparation were determined using two quantitative PCR assays45. Libraries were then amplified and double-indexed by PCR46. One library was prepared from each DNA fraction, except for the second and third 90 °C phosphate fractions from the DCP1 from which five libraries were prepared, each to maximize the yield of sequence data. Negative controls containing no sample material were carried along each set of extraction and library preparation. Note that there was substantial variation in library preparation efficiency across samples (Fig. 2 and Supplementary Data 1), ranging from 21.8% on average for the phosphate DNA fractions obtained from Bacho Kiro Cave specimens to 60.8% for DCP1 and 62.0% for the Quinçay specimens.
Enrichment of mitochondrial and nuclear DNA by hybridization capture
To determine the taxonomic composition of the DNA recovered from the artefacts, libraries prepared from nearly all extracts generated in this study (all samples and controls, with the exception of the second and third phosphate fractions from BKP3) were enriched for mammalian mtDNA by hybridization capture with a probe set (‘AA75’) encompassing the mtDNA genomes of 242 mammalian species24. In cases in which several libraries were prepared from the same DNA extracts (that is, the 90 °C phosphate fractions from DCP1), only the first library was enriched for mammalian mtDNA. In addition, all libraries were enriched specifically for hominin mtDNA using a probe set (‘AA163’) designed in 1-bp tiling based on the revised Cambridge reference sequence of the human mitochondrial genome32. Both types of mtDNA captures were performed using two consecutive rounds of automated capture as detailed in Slon et al. (2017)6 or Zavala et al. (2022)47. An overview of the capture reactions performed is provided in Supplementary Data 1.
In addition to mtDNA captures, 11 libraries prepared from the first, second and third 90 °C phosphate fractions of DCP1 were enriched for human nuclear DNA by two consecutive rounds of in-solution capture48. This enrichment was performed using a subset of a previously designed capture probe panel8, named AA204, targeting two groups of SNPs: (1) 59,232 SNPs, randomly selected from set 6 ‘hominin diagnostic sites’8, which represent positions in the genome where primates differ from other mammals and are used to quantify faunal mis-alignments; and (2) 411,492 SNPs selected from the ‘1240k’ panel30,49 (set 2 in Vernot et al. (2021)8). This set of SNPs is informative for investigating modern human population histories. Both SNP groups are located in regions of large evolutionary sequence divergence between humans and other mammals8.
Sequencing and raw sequence processing
Libraries enriched for mtDNA were combined into pools and sequenced on multiple lanes of a MiSeq sequencer (Illumina Technologies) in paired-end configuration with two index reads (2× 76 + 2× 8 cycles). Libraries enriched for human nuclear DNA were sequenced either on a HiSeq 2500 using the same configuration or on a HiSeq4000 (both Illumina Technologies) in single-read configuration with two index reads (1× 76 + 2× 8 cycles). In addition, one library prepared from the second 90 °C phosphate fraction of the DCP1 was sequenced directly (shotgun sequencing, without hybridization capture) on one lane of a HiSeq 4000 sequencer (Illumina Technologies) in single-read configuration. Base calling was performed using Illumina’s Bustard tool and sequences were assigned to the library that they originated from, requiring perfect matches to the expected index combinations. LeeHom (https://github.com/mpieva/leeHom/tree/v.1.1.5)50 was used to trim adapters and, for paired-end data, to merge overlapping paired-end reads.
Taxonomic assignment of mtDNA sequences
Sequences resulting from mammalian or human mtDNA capture were assigned to mammalian taxa on the biology family level using a previously published computational pipeline6 based on BLAST and MEGAN (version 0.0.12)51, with modifications in data filtering detailed in Vernot et al. (2021)8 (Supplementary Data 1 and Supplementary Information 4). In brief, as described in the latter study, false identifications of taxa were minimized by requiring at least three unique sequences (covering at least 105 positions in the reference genome) to be assigned to a family, and by requiring these sequences to represent at least 1% of all taxonomically identified sequences. The presence of ancient DNA was determined individually for the sequences from each family (and each library) by computing the frequency of terminal C-to-T substitutions as a proxy for the deamination rate at the molecule ends. Substitution frequencies significantly higher than 10% on both molecule ends (based on 95% binomial CIs) were then taken as evidence for the presence of ancient DNA from the respective family.
Human and cervid mtDNA analysis
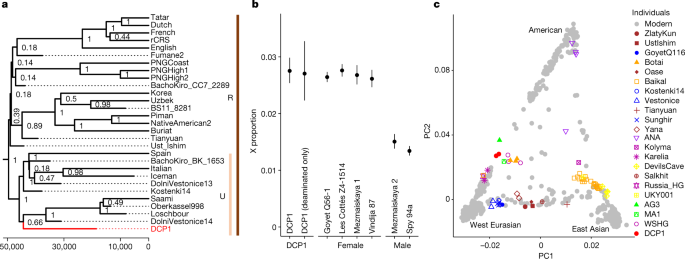
Present-day human contamination in the fractions from DCP1 that yielded ancient human mtDNA was estimated using the software tool AuthentiCT (version 1.0.0)52 and a near-complete consensus sequence called for the second 90 °C fraction from DCP1 using positions with at least tenfold coverage of the mtDNA genome. This consensus sequence was then used for haplogroup assignment with Haplogrep 2 (version2.4.0)53 and to identify ‘diagnostic’ positions in the mtDNA genome, which were used to determine the support for the consensus sequence in each of the DNA fractions recovered from DCP1 (Extended Data Table 3). Tree building and genetic dating were performed using BEAST2 (version 2.6.6)54. Further information on the mtDNA analyses are provided in Supplementary Information 5. For genetic dating of the deer DNA component, near-complete wapiti mtDNA genomes were reconstructed from the first 60 °C phosphate fraction obtained from DCP1, as well as eight ancient wapiti samples as described in detail in Supplementary Information 6.
Human nuclear DNA analysis
Human DNA capture data were processed as previously described8 and data from the libraries with the lowest estimates of modern human contamination (based on AuthentiCT) were merged for further analyses. Principal component analysis including sequence data from present-day and other ancient human individuals was performed using smartpca (from EIGENSOFT package version 8.0.0)55. Using the R package admixr (version 0.7.1)28, ƒ3-statistics were calculated to determine shared genetic drift between DCP1 and a selection of modern and ancient human populations and their relationships were further evaluated using D-statistics. Sex determination for the human DNA component recovered from DCP1 was performed by comparing the coverage of the X chromosome and the autosomes in shotgun data obtained from the second 90 °C phosphate fraction after filtering against faunal mis-mappings8. Further details are provided in Supplementary Information 7.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.